Infrared (IR) Spectroscopy
Infrared (IR) Spectroscopy is a powerful analytical technique used to identify functional groups in pure compounds based on their interaction with infrared radiation. The fundamental principle of IR spectroscopy involves the absorption of infrared light by molecules, leading to vibrational or rotational transitions.
Principle of Infrared Spectroscopy
-
Electromagnetic Radiation: Infrared radiation is a type of electromagnetic radiation with wavelengths ranging from approximately to (or frequencies between to ). Unlike ultraviolet (UV) spectroscopy, IR radiation does not have enough energy to induce electronic transitions but is suitable for exciting molecular vibrations.
-
Vibrational Transitions: When a molecule absorbs infrared radiation, it can undergo vibrational transitions. Molecules consist of atoms bonded together, and these bonds can stretch and bend. The specific vibrational modes depend on the type of atoms and the bonds within the molecule.
-
Dipole Moment Change: For a molecule to absorb IR radiation, there must be a change in the dipole moment during the vibrational motion. Molecules with a permanent dipole moment (like or ) are IR active because their vibration leads to changes in the dipole moment, allowing them to absorb IR radiation. In contrast, molecules that do not exhibit a change in dipole moment (like , , and ) are IR inactive and do not absorb IR radiation.
-
Frequency Matching: The frequency of the absorbed IR radiation must match the vibrational frequency of the molecule for absorption to occur. When this condition is satisfied, the molecule can effectively absorb energy, leading to a distinct spectral feature that can be analyzed.
-
Spectral Analysis: The resulting spectrum displays the intensity of absorbed light as a function of frequency or wavelength. Specific peaks in the spectrum correspond to different functional groups within the molecule, allowing for qualitative and quantitative analysis.
Types of IR Spectrum Region
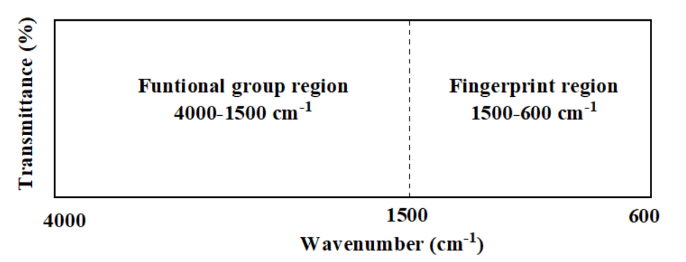
IR spectrum is divided mainly into two region as follows:
- The functional group region spans from 4000 cm⁻¹ to 1500 cm⁻¹. This part of the spectrum contains absorption bands that are characteristic of specific functional groups, such as hydroxyl groups (-OH), carbonyl groups (C=O), and amines (N-H). These vibrations are often simpler and associated with stretching of bonds between atoms, like C-H or O-H. Since the absorption bands for different functional groups appear in predictable ranges, this region is extremely useful for quickly identifying the presence of specific functional groups in a molecule. For example, the O-H stretch is commonly found between 3200 cm⁻¹ and 3600 cm⁻¹, while the C=O stretch appears around 1700 cm⁻¹ to 1750 cm⁻¹. This allows for rapid identification of certain molecular features.
- The fingerprint region covers the range from 1500 cm⁻¹ to 600 cm⁻¹. This region is more complex and contains numerous overlapping absorption bands, which arise from bending vibrations and stretches in the molecular backbone, such as C-C, C-N, or C-O bonds. Although this region is harder to interpret due to the complexity and overlap of vibrations, it is highly specific to each individual molecule. Much like a fingerprint, the pattern in this region is unique for different compounds. As a result, this part of the spectrum is used to confirm the identity of a substance by comparing its absorption pattern to known reference spectra. Small differences in molecular structure often result in noticeable changes in this region, making it very useful for distinguishing between similar compounds.
In practice, the functional group region is primarily used for identifying key functional groups, while the fingerprint region helps in confirming the unique identity of the molecule. Both regions are important for a complete understanding of the molecular structure when using infrared spectroscopy.
Instrumentation in IR Spectroscopy
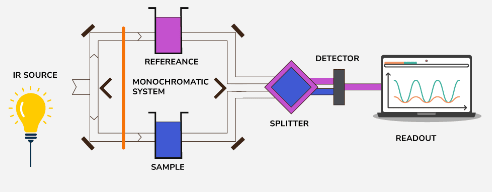
Instrumentation in IR spectroscopy typically involves the following components:
-
IR Source: A thermal source generates infrared radiation. In modern systems, this source may be a heated material like a Nernst glower or a Globar (silicon carbide). The IR light is emitted toward the monochromatic system.
-
Sample and Reference Holders: The system uses a dual-beam setup where IR light passes through both the sample chamber and the reference chamber. The sample holder contains the material being analyzed, while the reference contains a substance with no IR absorption to provide a baseline for comparison.
-
Monochromatic System: This component disperses the incoming IR radiation and selects the desired wavelength to pass through the sample. In modern systems, this is achieved using either prisms, gratings, or interferometers in FTIR instruments.
-
Beam Splitter: The IR beam is split, directing part of the light toward the sample and reference chambers and the other part toward the detector. This allows simultaneous measurement of the sample and reference.
-
Detector: After passing through the sample and reference, the IR radiation reaches the detector, which measures the intensity of transmitted radiation. Common detectors include thermocouples, pyroelectric detectors, or photoconductive detectors (e.g., MCT - mercury cadmium telluride).
-
Readout System: The data from the detector is sent to a readout system, typically a computer, where the absorption spectrum is displayed. The output shows peaks and troughs, corresponding to molecular vibrations.
-
Fourier Transform Infrared (FTIR) Spectroscopy: Modern IR spectrometers often use FTIR, which applies an interferometer rather than a monochromator. The interferometer collects all wavelengths at once, and the final spectrum is produced by applying a Fourier transformation. This technique is faster and provides higher resolution than traditional dispersive methods.
Fundamental Vibrations
Fundamental vibrations refer to the basic vibrational modes of a molecule that occur due to the stretching or bending of chemical bonds. These are the simplest and most energetically significant vibrations that can be observed in an infrared (IR) spectrum.
For a molecule with atoms, the total number of possible vibrational modes (both fundamental and non-fundamental) is given by:
- for non-linear molecules
- for linear molecules
These represent the independent ways in which the atoms in a molecule can vibrate.
Types of Fundamental Vibrations
-
Stretching Vibrations:
- Involve changes in bond length between atoms.
- There are two types:
- Symmetric Stretching: Atoms move simultaneously towards or away from each other in a coordinated manner.

- Asymmetric Stretching: One atom moves in while the other moves out, causing an imbalance in the stretching.

- Symmetric Stretching: Atoms move simultaneously towards or away from each other in a coordinated manner.
-
Bending Vibrations:
- Involve changes in bond angles rather than bond lengths.
- In plane bending vibrations:
- Scissoring: Both the atom move towards each other just like scissor.

- Rocking: Both the atoms move in same direction, either in left side or right side.

- Scissoring: Both the atom move towards each other just like scissor.
- Out of plane bending vibrations:
- Wagging: Both the atom move up and down with respect to central atom.

- Twisting: One atom move up and other atom move down with respect to central atom.

- Wagging: Both the atom move up and down with respect to central atom.
Visualizing the Vibrations
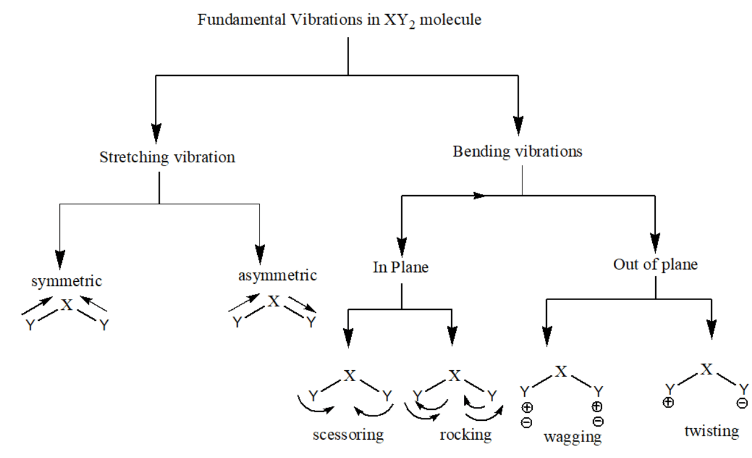
- Stretching occurs along the axis of the bond, altering the bond length.
- Bending occurs perpendicular to the bond axis, altering the bond angles.
Examples
-
Water (H₂O): Water is a non-linear molecule, and its fundamental vibrations include:
- Symmetric stretching (~3650 cm⁻¹)
- Asymmetric stretching (~3750 cm⁻¹)
- Bending (~1600 cm⁻¹)
-
Carbon Dioxide (CO₂): Although linear, CO₂ has:
- Symmetric stretching (IR inactive)
- Asymmetric stretching (~2349 cm⁻¹)
- Bending (scissoring) (~667 cm⁻¹)
Factors Affecting Vibrational Frequency
-
Conjugation: Conjugation refers to the delocalization of π-electrons across adjacent double bonds or lone pairs. When conjugation occurs, the force constant (k) of the bond decreases because the bond becomes less localized, effectively weakening it. As a result, the stretching frequency decreases.

The left structure is a simple ketone with a C=O stretching frequency at 1720 cm⁻¹. The right structure shows a conjugated system, where the C=O bond is adjacent to a C=C double bond (alkene). Due to this conjugation, the C=O stretching frequency decreases to 1680 cm⁻¹. This happens because conjugation lowers the force constant of the carbonyl bond (C=O) by delocalizing the electron density, thus weakening the bond and decreasing the stretching frequency. This agrees with the principle that as conjugation increases, the stretching frequency decreases.
-
Nature of Substituents:
-
Inductive Effect: Electronegative atoms or groups (like oxygen) withdraw electron density from nearby bonds, thereby increasing the bond strength. For example, in a carbonyl group, if an electronegative atom like oxygen is attached to the molecule (e.g., in an ester), it increases the stretching frequency of the C=O bond due to the withdrawal of electrons through the inductive effect.
-
Resonance Effect: In contrast, the resonance effect tends to decrease the stretching frequency by delocalizing electron density. For example, when the lone pair of nitrogen in an amide (R-C=O-NH₂) delocalizes into the carbonyl group, the C=O bond partially loses its double bond character. This reduces the force constant and lowers the stretching frequency.

- Top Half:
-
Ester (C=O attached to -OR group):
- The C=O stretching frequency is shown as 1735 cm⁻¹, which is slightly higher due to the -I (inductive) effect of the -OR group. The electronegative oxygen atom in the -OR group withdraws electron density from the C=O bond, increasing the bond's strength and thus increasing the stretching frequency.
-
Amide (C=O attached to -NH₂ group):
- The C=O stretching frequency is shown as 1690 cm⁻¹, which is lower than that of the ester. This is due to the +R (resonance) effect of the -NH₂ group. The lone pair of nitrogen can delocalize into the C=O bond, reducing the bond's double-bond character and decreasing the stretching frequency.
- Bottom Half:
-
Simple Ketone (H₃C-CO-CH₃):
- The C=O stretching frequency is 1720 cm⁻¹, which reflects the normal stretching frequency without significant inductive or resonance effects.
-
Ketone with CF₃ group (H₃C-CO-CF₃):
- The C=O stretching frequency increases to 1760 cm⁻¹ due to the -I (inductive) effect of the electron-withdrawing CF₃ group. This effect pulls electron density away from the carbonyl group, strengthening the bond and raising the frequency.
-
Trifluoromethyl Ketone (F₃C-CO-CF₃):
- The C=O stretching frequency further increases to 1790 cm⁻¹ due to the strong -I (inductive) effect of two CF₃ groups. The strong electron-withdrawing nature of the CF₃ groups further increases the bond strength, leading to a higher stretching frequency.
-
-
Hydrogen Bonding:
- Intermolecular Hydrogen Bonding: This type of bonding weakens the bond being studied (like O-H or N-H bonds) by creating additional interactions with neighboring molecules. This lowers the stretching frequency. In the case of alcohols (R-OH), hydrogen bonding in concentrated solutions causes the O-H stretching frequency to shift to lower values (e.g., from ~3600 cm⁻¹ in a dilute solution to ~3300 cm⁻¹ in a concentrated solution).
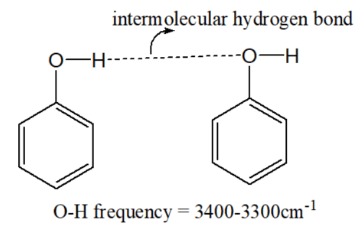
- Intramolecular Hydrogen Bonding: Intramolecular hydrogen bonds (within the same molecule) also decrease the stretching frequency, but these are more rigid and stable. For example, in methyl salicylate, strong intramolecular hydrogen bonding lowers the O-H stretching frequency to ~3200 cm⁻¹, and this value remains constant even in dilute solutions because the molecular structure does not change with dilution.

- Intermolecular Hydrogen Bonding: This type of bonding weakens the bond being studied (like O-H or N-H bonds) by creating additional interactions with neighboring molecules. This lowers the stretching frequency. In the case of alcohols (R-OH), hydrogen bonding in concentrated solutions causes the O-H stretching frequency to shift to lower values (e.g., from ~3600 cm⁻¹ in a dilute solution to ~3300 cm⁻¹ in a concentrated solution).
-
Ring Strain: As the size of a cyclic ring decreases, the bond angles become more constrained, and the C=O bond becomes more difficult to stretch. This increase in rigidity causes the vibrational frequency of the C=O bond to increase.

In cyclic ketones, the carbonyl (C=O) stretching frequency increases as the ring size decreases due to ring strain. Here's how it works:
- Larger rings (e.g., cyclohexanone, left): The carbonyl stretching frequency is around 1710 cm⁻¹ because the bond angles are closer to their ideal values, leading to less strain.
- Medium rings (e.g., cyclopentanone, middle): The carbonyl stretching frequency increases to 1745 cm⁻¹ due to a moderate amount of ring strain. The bond angles deviate more from the ideal, and the increased strain raises the C=O bond vibrational frequency.
- Smaller rings (e.g., cyclobutanone, right): The stretching frequency is highest at 1780 cm⁻¹. This is because the bond angles are very compressed in a four-membered ring, creating significant strain that makes the C=O bond harder to stretch, leading to an increase in vibrational frequency.
Hooke's Law
In Infrared (IR) Spectroscopy, Hooke's Law models the bond between two atoms as a spring. The frequency of the bond's vibration is related to the force constant of the bond (which represents bond strength) and the reduced mass of the atoms involved. The basic equation is derived from Hooke’s Law:
Where:
- = vibrational frequency
- = wavenumber (cm⁻¹), which is proportional to the frequency
- = force constant of the bond (strength of the bond)
- = reduced mass of the two atoms connected by the bond
- = speed of light
- , where and are the masses of the two atoms involved.
Effect of Masses of Atoms
- The reduced mass () is inversely proportional to the frequency of vibration. Lighter atoms (with lower masses) result in a higher frequency of vibration because the bond can vibrate more quickly.
- For example, bonds involving hydrogen (lighter atom) will generally vibrate at a higher frequency compared to bonds involving heavier atoms like carbon or oxygen.
Effect of Bond Strength (Force Constant)
- The stronger the bond, the higher the force constant (). Stronger bonds are harder to stretch, so they vibrate at higher frequencies.
- For example:
- C-C bond has a lower force constant () and vibrates at 1200 cm⁻¹.
- C=C bond has a higher force constant () and vibrates at 1650 cm⁻¹.
- C≡C bond has the highest force constant () and vibrates at 2100 cm⁻¹.
Applications of IR spectroscopy
-
Identification of different functional groups: IR spectroscopy is widely used to identify the presence of various functional groups in a molecule (e.g., C=O, O-H, N-H). Each functional group absorbs IR radiation at specific frequencies, allowing for their identification.
-
Distinction between intermolecular and intramolecular hydrogen bonding: IR spectroscopy can help differentiate between types of hydrogen bonding. Intermolecular hydrogen bonding (between different molecules) and intramolecular hydrogen bonding (within the same molecule) lead to different shifts in IR absorption peaks.
-
Identification of purity of a compound: IR spectroscopy can be used to assess the purity of a sample. If a compound contains impurities, additional peaks corresponding to different molecular structures or functional groups will appear in the IR spectrum, indicating the presence of contaminants.
-
Study of chemical reactions: IR spectroscopy can track changes in a compound during a chemical reaction. For example, in the image, the reduction of a carbonyl group (C=O) in a compound is followed by the appearance of an alcohol (O-H) group, which can be detected by the shifts in the IR spectrum (from 1710 cm⁻¹ for the C=O to 3300 cm⁻¹ for the O-H).

-
Identification of geometrical isomers (cis-trans): IR spectroscopy is useful in distinguishing between different geometrical isomers, such as cis-trans isomers, based on the differences in their IR absorption patterns due to the spatial arrangement of the atoms.
UV-Visible Spectroscopy
UV-Visible Spectroscopy-principle, Lambert-Beer's Law, instrumentation, Electronic Transitions, Auxochromes, Chromophores, Effect of conjugation and solvents on transition of organic molecules, applications.
Fuels
Classification of fuels, Calorific value - Definition, HCV, LCV, determination of calorific value of solid and liquid fuels using Bomb calorimeter, Ultimate analysis of coal and numerical problems, Petroleum cracking -fluidized bed catalytic cracking. Reformation of petrol, Quality of liquid fuels- Cetane and Octane number, power alcohol-manufacture, advantages and disadvantages, Concept of hydrogen as fuel- types, synthesis by water electrolysis and natural gas reforming.